Functions and Data Types
Outline
Review: Write yourself some functions
Types: How they matter and how to avoid being bitten by them
Function practice: Normalizing RNA-Sequencing
We will be using part of the data from a study on spinal motor neurons. You can find their full analysis scripts here.
# Use read.delim instead of read.csv for **tab**-delimited files
rawcount = read.delim("BR-A-Control_counts.txt")What is in the raw data?
# Inspect the data you load!normalize_by_depth()
How do we define a function that normalize a vector of counts by total counts?
Before you start writing. Let’s start humble and get a small proportion of the data so you can capture errors right away when you test your function.
The key of doing this is to have something that you can tell whether your code is doing something you want right away with test data structurally similar to the real data.
# Take a small fraction of the data so we can test our function
test_count = head(rawcount)Now, we are good to go. Let’s get the total count first!
# What is our input?
normalize_by_depth = function(input) {
# How do we compute total count?
# Let's divide everything by the total count calculated above
# Provide an output
}Let’s test the function with our naked eyes.
# Is the function doing what we expect it to do?
normalize_by_depth(test_count) == 1472 + 6 + 109 + 43## logical(0)If everything goes as expected, let’s divide everything by total count in the function, too.
# What is our input?
normalize_by_depth = function(input) {
# How do we compute total count?
depth = sum(input$count)
# Let's divide everything by the total count calculated above
# Provide an output
# **Note that you would want to change your output!**
return(depth)
}Let’s test the revised function. What are we expecting here?
# We are expecting the first gene to be ~0.903 after normalization
# while the second gene is 0.
# Let's run the function on the test set and let the results print out.Let’s multiply the result by \(10^{6}\) (10^6) before returning it in our function.
# What is our input?
normalize_by_depth = function(input) {
# How do we compute total count?
depth = sum(input$count)
# Let's divide everything by the total count calculated above
normalized_count = input$count/depth
input$normalized_count = normalized_count
# Provide an output
return(input)
}And test it again.
Note that for the first gene, we used to getting 0.9030675 before multiplying with \(10^{6}\).
normalize_by_depth(test_count)## id count normalized_count
## 1 ENSMUSG00000000001 1472 0.903067485
## 2 ENSMUSG00000000003 0 0.000000000
## 3 ENSMUSG00000000028 6 0.003680982
## 4 ENSMUSG00000000031 109 0.066871166
## 5 ENSMUSG00000000037 43 0.026380368
## 6 ENSMUSG00000000049 0 0.000000000get_tx_length()
A gene model file contains the starting and ending coordinates of genes, transcripts, and exons.
You can get a gene model file from Ensembl.
They are often stored as GTF/GFF3 files, but the format is beyond the scope of what we are going to do today. If you are interested, you can find more information about GTF/GFF format here.
# Load gene model file (pre-processed)
gene_model = read.csv("mouse_gene_model.csv")
head(gene_model)## id start end
## 1 ENSMUSG00000102628 150956201 150958296
## 2 ENSMUSG00000100595 150983666 150984611
## 3 ENSMUSG00000097426 151012258 151012971
## 4 ENSMUSG00000097426 151013347 151013531
## 5 ENSMUSG00000104478 108344807 108347562
## 6 ENSMUSG00000104385 6980784 6981446How do we define a function that get us lengths for each gene?
–
library(dplyr)##
## Attaching package: 'dplyr'## The following objects are masked from 'package:stats':
##
## filter, lag## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union# How do we compute the length of each exons?
gene_model %>%
head() %>%
mutate(length = (end - start + 1))## id start end length
## 1 ENSMUSG00000102628 150956201 150958296 2096
## 2 ENSMUSG00000100595 150983666 150984611 946
## 3 ENSMUSG00000097426 151012258 151012971 714
## 4 ENSMUSG00000097426 151013347 151013531 185
## 5 ENSMUSG00000104478 108344807 108347562 2756
## 6 ENSMUSG00000104385 6980784 6981446 663Let’s test if the length column is correctly calculated.
# For the first gene
150958296 - 150956201 + 1## [1] 2096If it seems right, let’s get the sum of lengths per id now.
get_tx_length = function(input) {
# How do we compute the sum of all exons of a gene?
tx_length = input %>%
mutate(length = (end - start + 1))
# Group
# Summarize by summation
# Provide an output
return(tx_length)
}What does the output look like?
gene_model %>%
head() %>%
get_tx_length()## id start end length
## 1 ENSMUSG00000102628 150956201 150958296 2096
## 2 ENSMUSG00000100595 150983666 150984611 946
## 3 ENSMUSG00000097426 151012258 151012971 714
## 4 ENSMUSG00000097426 151013347 151013531 185
## 5 ENSMUSG00000104478 108344807 108347562 2756
## 6 ENSMUSG00000104385 6980784 6981446 663How do you independently test if the answer is correct for ENSMUSG00000097426?
# Doing it differently with base R
gene_of_interest = gene_model[gene_model$id == "ENSMUSG00000097426", ]
print(gene_of_interest)## id start end
## 3 ENSMUSG00000097426 151012258 151012971
## 4 ENSMUSG00000097426 151013347 151013531gene_of_interest$length = gene_of_interest$end - gene_of_interest$start + 1
print(gene_of_interest)## id start end length
## 3 ENSMUSG00000097426 151012258 151012971 714
## 4 ENSMUSG00000097426 151013347 151013531 185sum(gene_of_interest$length)## [1] 899One function to do it all
You must have noticed that functions are like Russian dolls: There are always functions inside a function.
Now that we have normalize_by_depth to generate CPM, get_tx_length to
calculate gene length, and we know that left_join can merge them by ID, we
can write a master function to streamline everything.
normalize_rnaseq = function(count, gene_model){
# 1. Normalize read counts by sequencing depth = total reads we got from a sample.
# (This gives CPM)
# 2. Normalize again with transcript length.
# (CPM/Gene length = Transcript per million (TPM))
# 3. Make a master table containing both CPM and length per gene
return(normalized_count)
}Finally, let’s test it:
normalize_rnaseq(test_count, test_gene_model)## Error in normalize_rnaseq(test_count, test_gene_model): could not find function "normalize_rnaseq"All your hard work pays now – you can normalize the whole thing with ease!
# Normalize the full table with full gene modelCommon pitfalls on data types
characters are friendly most of the time, but…
Alphabetical and numerical sorting
chr_vec = c("5", "8", "6", "10", "7", "9")# You might not expect it to sort like this
sort(chr_vec)## [1] "10" "5" "6" "7" "8" "9"# If they are numbers, they sort differently
sort(as.numeric(chr_vec))## [1] 5 6 7 8 9 10Numeric type: Precision can be dangerous…
0.1 + 0.2 == 0.3## [1] FALSEThere’s a website called https://0.30000000000000004.com/ that explains this in detail.
But briefly, any number that is not an integer has limited precision, and propagation of error is a thing.
# A more robust way to compare non-integers
# Define an error margin that you want to tolerate
error_margin = 10^-8
# and then decide if the difference is within the margin
(0.1 + 0.2) - 0.3 < error_margin## [1] TRUEFactor: Ordered categories
# Categories as characters works most of the time, but...
month_tbl = data.frame(
month = c(
"January", "February", "March", "April", "May", "June",
"July", "August", "September", "October", "November", "December"
),
length = c(
31, 28, 31, 30, 31, 30, 31, 31, 30, 31, 30, 31
)
)
str(month_tbl)## 'data.frame': 12 obs. of 2 variables:
## $ month : chr "January" "February" "March" "April" ...
## $ length: num 31 28 31 30 31 30 31 31 30 31 ...Most functions that you encounter in R will sort alphabetically for characters.
library(ggplot2)
month_tbl %>%
qplot(data = ., x = month, y = length, geom = "point") +
# This adjust the axis text to make the text more visible
theme(axis.text.x = element_text(size = 20, angle = 60, hjust = 1))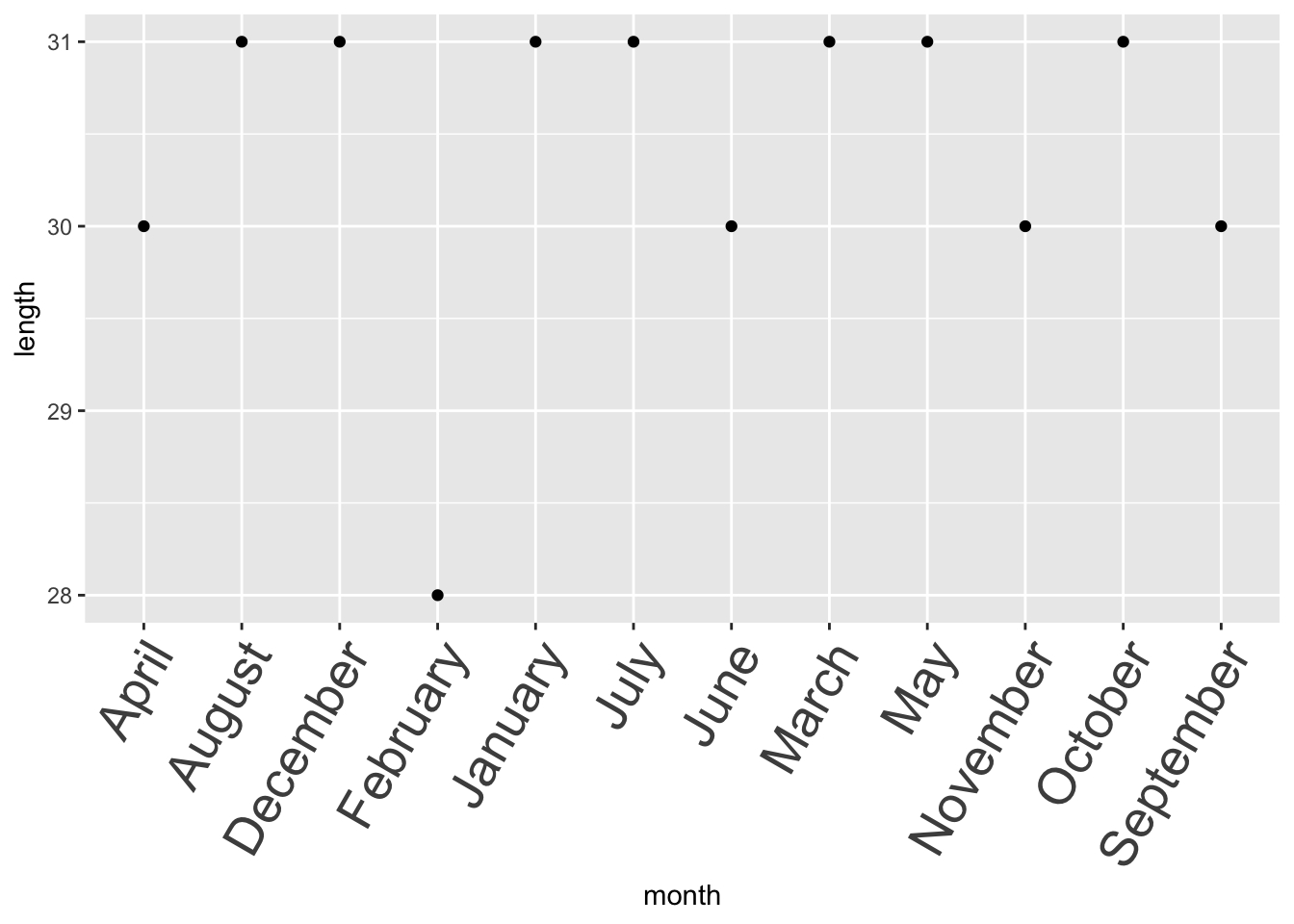
You won’t be able to decide the order unless you convert it to a factor.
month_tbl$month = factor(
month_tbl$month,
# R will respect the levels you set here
levels = c(
"January", "February", "March", "April", "May", "June",
"July", "August", "September", "October", "November", "December"
)
)
month_tbl %>%
qplot(data = ., x = month, y = length, geom = "point") +
# This adjust the axis text to make the text more visible
theme(axis.text.x = element_text(size = 20, angle = 60, hjust = 1))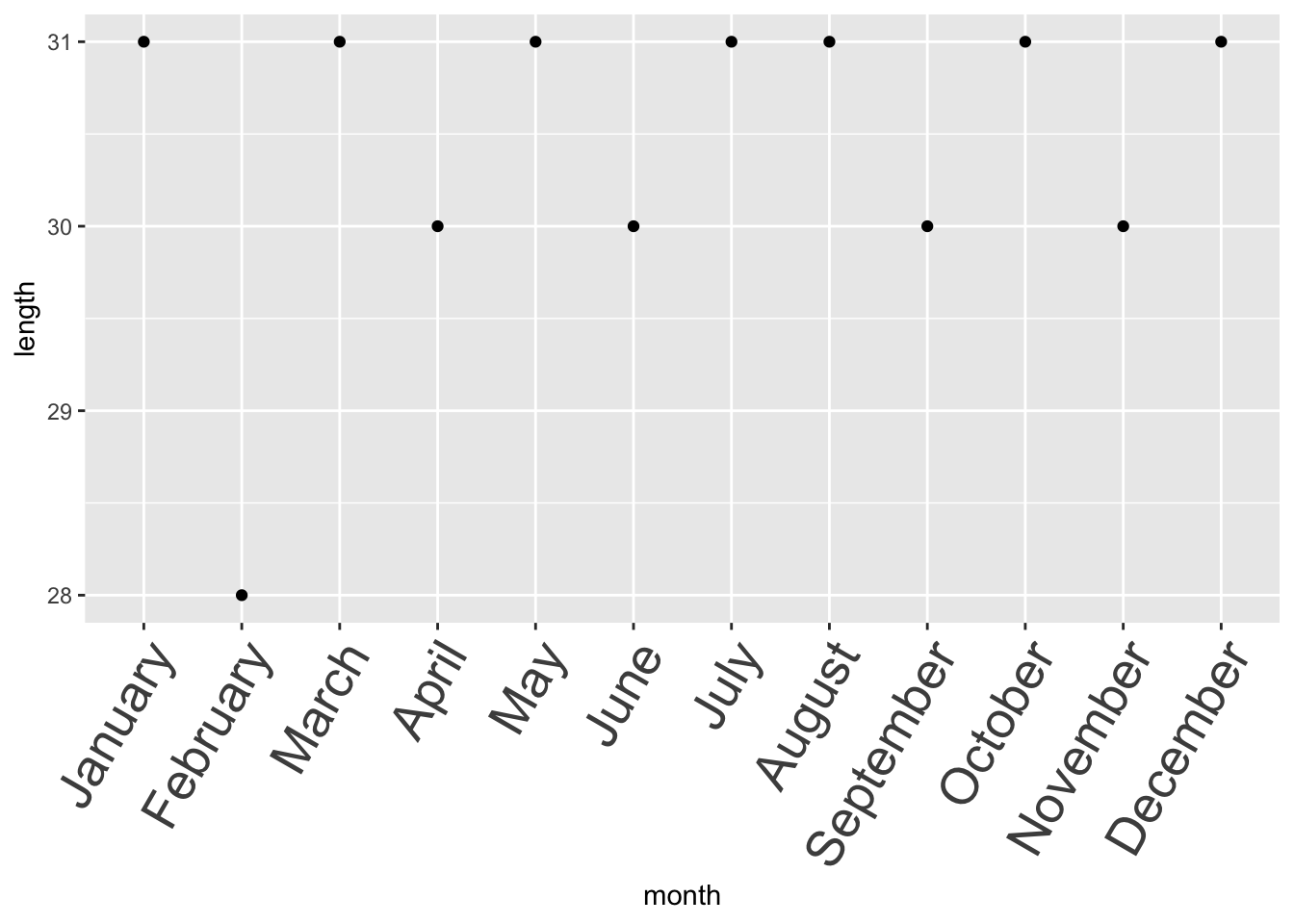
Factors are more complicated than characters and numbers, so they could be harder to troubleshoot, but at the same time, they are very powerful especially in statistics.
As a rule of thumb, when you inspect your data (e.g., with str()), always ask
yourself if you are having categorical variables.
If you do, consider converting them to factors if:
You know they are ordinal
When you are building statistical models with categorical variables (ANOVA et al.)
Date and time
They have a similar issue as ordinal categories: If considered as characters, they won’t be ordered chronologically.
# This shouldn't be surprising by now
random_dates = c("12-25-2022", "07-04-1989", "01-01-2077")
sort(random_dates)## [1] "01-01-2077" "07-04-1989" "12-25-2022"Unlike most categorical variables, there are usually so many dates, so it is not practical for you to assign orders manually.
Luckily, there is a package that will take care of this for you if you tell it the format of your dates.
# You can tell R how the date is represented to chronologically sort
library(lubridate)##
## Attaching package: 'lubridate'## The following objects are masked from 'package:base':
##
## date, intersect, setdiff, union# mdy() stands for month, day, year
good_dates = mdy(random_dates)
sort(good_dates)## [1] "1989-07-04" "2022-12-25" "2077-01-01"Yen-Chung Chen
PhD Candidate
A graduate student interested in developmental biology, neurobiology and bioinformatics.